CEP/CER White Paper: Launching Class I Medical Devices in the UK and EU Markets
Navigating Regulatory pathways for launching Class I Medical Devices in the UK and EU Markets
This white paper, presented by Cormica, a global leader in regulatory and laboratory testing services, outlines the strategic approach for launching Class I devices in the UK and EU.
Cormica’s laboratories have over 60 years of experience and global regulatory expertise, supporting Class I medical device manufacturers in navigating regulatory pathways from technical documentation through post-market surveillance
Class I medical devices encompass non-invasive, low-risk products such as bandages, wheelchairs, and reusable surgical instruments. While the regulatory process is more straightforward than higher-risk categories, manufacturers must adhere to essential requirements to ensure market success.
Understanding the specific pathways for UK and EU regulatory approval and ongoing compliance measures is crucial.
Medical device manufacturers launch their medical devices on the EU and UK markets to grab the ongoing demand for medical devices in the medical field. After the pandemic effect, the manufacturers are proactively supplying the medical devices to the distribution centers or subsidiaries to maintain the supply chain management.
With an ever-growing demand for medical devices, there is still a need for more supply chain management. Newly started-up medical device or non-EU/UK manufacturers need to understand how to get CE and UKCA labeling to market the products.
Regulatory requirements are applicable based on the route the manufacturer wants to launch the medical devices. Failure to meet these requirements can result in delays or even rejection of products, highlighting the importance of thorough research and planning before entering new markets.
Read on for further detail
Type:
Published:

Uday Kuppam
Uday Praveen Kuppam is a Clinical Evaluation Specialist with 10+ years’ experience in CERs, PSURs, BERs, and EU MDR compliance across multiple therapeutic areas.
Regulatory Pathway for Class I Medical Devices
1. Compliance with EU MDR (2017/745)
- Class I devices must conform to the General Safety and Performance Requirements (GSPR).
- Manufacturers must prepare a Technical File, including product descriptions, intended use, risk assessment, and labelling.
- Self-certification applies unless the device has a measuring function or is sterile (Class Is or Im), which requires a Notified Body assessment.
- CE marking is required for market entry in the EU.
2. Compliance with UK MDR (2002, as amended)
- Post-Brexit, UK regulatory requirements differ slightly from the EU.
- Class I devices require self-certification for UKCA marking.
- UK Responsible Person (UKRP) is mandatory for non-UK manufacturers.
- Device registration with the Medicines and Healthcare products Regulatory Agency (MHRA) is required.
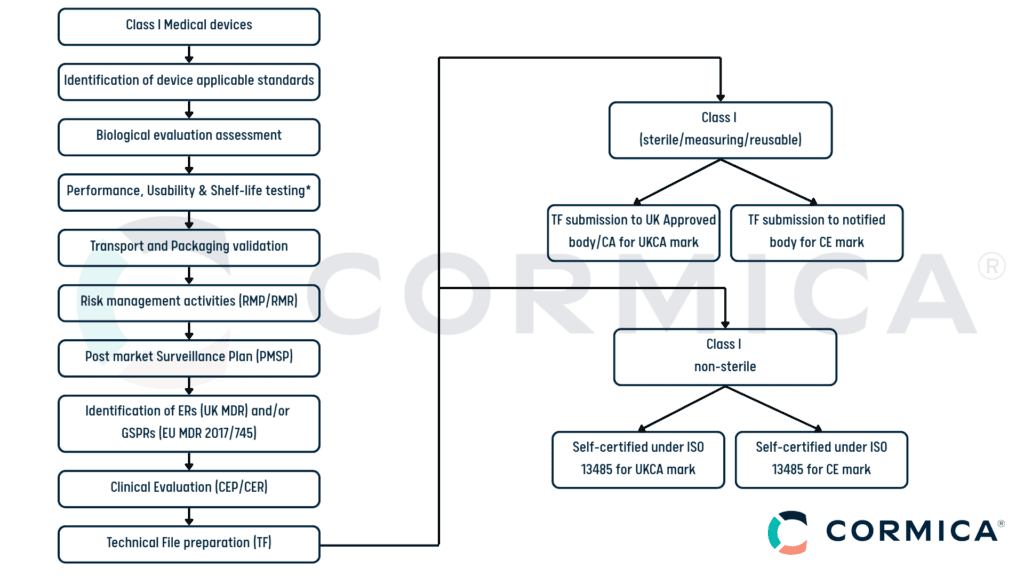
Note*: Sterility testing/validation is required for the sterile devices.
Other Regulatory Pathways to Place The Devices on the UK Market
For EU manufacturers:
According to translational arrangement, legislation has been introduced to extend when CE-marked devices may be placed on the Great Britain market. Medical devices CE marked under MDD are accepted up to 30th Jun 2028, and CE marked under MDR are accepted up to 30th Jun 2030 to place on the UK market. After 2028 or 2030, CE-marked devices are placed on the UK market through the international reliance route.
For non-UK manufacturers:
Currently, submission of technical files to the UK-approved body for UKCA labelling is undergoing. However, MHRA introduced a proposal of an international reliance route for non-UK manufacturers, currently seeking public and expert opinion, which may be in place after 2028. Based on the proposal, low-risk devices (Class I non-sterile medical devices) that comply with devices legislation in Australia, Canada, the EU, and the USA are allowed through route 1.
That means no approved body review is required, and a self-declaration for an appropriate Quality Management System (QMS) must be provided to the MHRA during registration. For Class Is/1m devices, approved body review is required, which is Route 3.
This proposed international reliance route aims to streamline the process for certain low-risk medical devices, reducing the burden on manufacturers. It is important for manufacturers to stay informed about any updates or changes to regulations to ensure compliance with UKCA labelling requirements.


Key strategies:
Regulatory preparation and Compliance
- Conduct a thorough gap analysis to ensure compliance with both UK and EU regulations.
- Maintain a comprehensive Technical File and Declaration of Conformity.
- Engage with regulatory consultants or experts to navigate complex requirements specifically for clinical evaluation and technical file preparation.
Post-Market Surveillance (PMS) and Vigilance
- Implement a robust PMS plan, including periodic safety updates and incident reporting.
- Engage with healthcare professionals and users for real-world performance insights.
- Ensure continuous compliance with MDR and UK MDR amendments.
Conclusion:
The successful launching of Class I medical devices in the UK and EU markets requires a strategic approach through early regulatory engagement and efficient documentation. As regulatory landscapes evolve, manufacturers must remain agile and proactive in addressing compliance challenges. This includes staying up to date on changing regulations, conducting thorough risk assessments, and ensuring all necessary documentation is in place before submitting for approval. By taking a proactive approach and working closely with regulatory authorities, manufacturers can streamline the approval process and minimise delays in bringing their products to market.
Cormica supports clients across all these stages, offering GMP, GLP and ISO 17025 – accredited laboratories across the UK, US and EU. Our team of regulatory and scientific experts ensures your Class I medical devices meet regulatory expectations, first time right.
References:
1. European Union Medical Device Regulation (EU MDR) 2017/745
2. UK Medical Device Regulations (UK MDR) 2002 (as amended)
3. Medicines and Healthcare products Regulatory Agency (MHRA) Guidance
4. European Commission Medical Devices Sector
5. Implementation of the future regulations – GOV.UK
6. Medical Devices Regulations: Routes to market and in vitro diagnostic devices